Alzheimer's Disease Through the Lens of Energy
Economics: Adaptive Origins, Comparative Clues & Therapeutic Opportunities
Jared
Edward Reser Ph.D.
1. Introduction: From Toxic Debris to Energy Economics
1.1
The metabolic-reduction hypothesis (Reser 2009): core logic
In
2009 I published an article proposing that Alzheimer's pathology may represent
a maladaptive extension of an evolutionarily conserved brain-energy reduction
mechanism. Central to the hypothesis is the idea that the human brain poses a
substantial energetic liability, especially in older hunter-gatherers facing
diminished foraging returns and resource availability. The article argued that
Alzheimer's would have selectively tempered the most energy demanding areas of
the brain to reduce overall energy expenditure without compromising foraging
ability. However, when modern longevity lets that same program run far
longer than hunter gatherers would have lived in the prehistoric past (55 is
the average terminal age of modern hunter-gatherers), it crosses the threshold
from adaptive thrift to clinical Alzheimer’s.
From
an evolutionary standpoint, humans historically experienced significant
reductions in caloric returns starting around age 45 to 50, when cognitive
demands for novel learning typically diminish because of accumulated expertise.
I argued that natural selection favored mechanisms enabling late-life
individuals to strategically reduce cortical energy expenditure by selectively
pruning metabolically expensive neuronal circuits, especially within high-order
association cortices and the hippocampus. Such an adaptive "low-power
mode" would have conserved valuable metabolic resources during prolonged
periods of scarcity without severely compromising essential survival skills,
sensorimotor functions, or culturally valuable accumulated knowledge. This dovetails
with other efforts made throughout the body to conserve energy with age. It is
also in line with the fact that brain metabolic rate diminishes linearly after
late childhood.
I
further noted striking parallels between the molecular and cellular hallmarks
of Alzheimer’s—such as selective regional glucose hypometabolism, synapse
elimination, insulin resistance, amyloid accumulation, and tau
hyperphosphorylation—and those observed in mammalian brains undergoing adaptive
metabolic downshifts. These downshifts include conditions such as starvation,
torpor, and hibernation. Intriguingly, these extreme yet reversible
physiological states share many molecular mechanisms, suggesting that
Alzheimer's pathology might represent a chronic activation of an ancient
metabolic conservation strategy. Before we continue, let us look at the key
lines of evidence from the 2009 article.
|
Evidence
category |
Core
point in the paper |
|
1. Human brain is an extreme
metabolic liability |
The cortex consumes 20‑25 % of
resting energy—more than any other organ—so trimming its cost in late life
would have increased survival during food scarcity. |
|
2. Age‑linked cerebral
hypometabolism is universal and continuous |
Brain glucose use peaks in
childhood, declines in adulthood, and keeps falling through old age; AD
represents a natural continuation of this trajectory. |
|
3. Selective hit‑map matches an
energy‑saving strategy |
AD preferentially deactivates high‑cost
association cortex & hippocampus while sparing sensorimotor areas;
exactly the pattern expected if the goal is to drop “expendable” cognitive
load yet preserve basic perception and movement. |
|
4. Parallels with starvation /
torpor responses |
Hyper‑phosphorylated tau, lowered
thyroid & growth hormone, insulin resistance, etc., appear in starving and hibernating mammals, and in AD, implying a shared fuel‑deprivation
program. |
|
5. Life‑history logic: declining
foraging & rising expertise |
Hunter‑gatherers’ calorie return
is reduced after around 45, by then skills are routinized, so costly working‑memory
circuits can be pruned without loss of competence. |
|
6. Metabolic‑syndrome &
“thrifty” genetics link |
AD co‑occurs with low resting
metabolic rate, insulin resistance, APOE‑ε4 and other thrifty alleles; their
geographic distribution tracks famine‑prone populations. |
|
7. Neuroecology parallels in other
species |
Some birds & mammals down‑regulate
hippocampal metabolism when possible; hippocampus is likewise the
earliest, most hypometabolic region in AD. |
|
8. Cross‑species ubiquity of
plaques & tangles |
Multiple mammalian orders show AD‑like
lesions, implying deep evolutionary roots and selective value rather than
random human pathology. |
Table 1: Key lines of evidence in the 2009 Reser article. Because the ageing brain’s energy budget
once exceeded what late‑life foragers could reliably earn, natural selection
favoured a program that (i) progressively
lowers cerebral metabolism, (ii) targets the most
expendable cognitive networks, (iii) mirrors documented
starvation/torpor responses, and (iv) is embedded in
thrifty genes that remain common wherever famine shaped human evolution.
The main thrust of this argument can be understood by relating it to two core concepts in the psychology of learning. I believe that Alzheimer’s disease can be reframed as a shift in the balance between two fundamental modes of learning that Jean Piaget described: assimilation and accommodation. Assimilation is the process of fitting new experiences into existing schemas, while accommodation is the restructuring of those schemas when they no longer match reality. In youth and adulthood both are essential, as the brain is still building, testing, and reshaping its store of knowledge. But by middle age, much of this work has already been done. The great lessons of life, how to survive, navigate relationships, and interpret cultural norms, have been internalized. At that point, the constant restructuring of knowledge becomes less critical, while the interpretive use of what is already known remains valuable. Thus, the memory consolidation and synaptic plasticity linked to accommodation may decline because the “return on investment” for constant schema-updating is low.


1.2 Purpose
and scope of this review
Over
the past fifteen years, accumulating data from human neuroimaging studies,
comparative biology, genetics, and clinical metabolic interventions have
provided support for the 2009 proposition. The purpose of this review is to
comprehensively synthesize and critically assess recent evidence that frames
Alzheimer's disease as an over-extended brain energy-conservation response
rather than purely a toxic amyloid-driven pathology. Vast overlap between
Alzheimer's and energy conservation modes will be documented, looking carefully
at similarities in molecular pathways with mammalian hibernation, torpor, and
starvation. The article will also examine translational therapeutic
opportunities informed by this reconceptualization of AD. By leveraging evolutionary insights and comparative biological
data, I propose novel therapeutic avenues designed not just to slow the
progression of AD, but to potentially reverse pathological changes by
re-engaging the brain’s inherent metabolic "wake-up" mechanisms.

Before we continue, let me spell out some of the
major similarities between Alzheimer's and adaptive metabolism reduction
programs in mammals in a didactic, easy-to-understand way.
As we continue, let me
spell out some of the major similarities between Alzheimer's and adaptive
metabolism reduction programs in mammals in a didactic, easy-to-understand
way.
2. Comparative Review:
Starving and Hibernating Mammals Utilize the Molecular Machinery of Alzheimer's
2.0
Hibernation and Torpor Explained
Many
animals face seasonal periods of food scarcity, particularly during winter
months when foraging becomes energetically costly or ecologically unfeasible.
To survive these conditions, certain species have evolved physiological
strategies that dramatically lower their metabolic demands. Hibernation is
one such strategy, characterized by prolonged, uninterrupted periods of
metabolic suppression. During hibernation, animals such as bears, bats, ground
squirrels, and groundhogs exhibit profound reductions in heart rate, body
temperature, and neural activity. For instance, hibernating bears may reduce
their metabolic rate by as much as 50%, entering a state of sustained energy
conservation.
Torpor,
by contrast, is a shorter-term, often daily or intermittent form of metabolic
depression, observed in species like skunks, chipmunks, hedgehogs, raccoons,
and deer mice. Torpor is also found across other taxa, including various birds,
reptiles, amphibians, and insects. While hibernation is typically seasonal and
extended, torpor allows for rapid, flexible responses to acute environmental
stressors such as cold or food shortage. Both hibernation and torpor, along
with the broader mammalian response to starvation, serve the adaptive function
of minimizing energy expenditure under resource-limited conditions. For
context, sleep also offers a mild energy-conserving effect—lowering
respiratory rate, body temperature, and metabolic rate—though unlike
hibernation and torpor, brain activity during sleep remains relatively high and
structured, reflecting its distinct functional role. The relation between AD
and brumation, estivation, and lethargy is also ripe for study.
2.1
Hibernators and Starvation: Reversible Tau and Synapse Stripping
During
deep hibernation, species such as ground squirrels, hamsters, and bears
dramatically reduce their brain's metabolic rate—sometimes by up to
90%—accompanied by extensive tau hyperphosphorylation at the very same
epitopes (AT8, Ser396) observed in human Alzheimer's brains
(Arendt et al., 2003; Stieler et al., 2011). In parallel, dendritic spine
density and synaptic connections are substantially pruned, reducing the
metabolic load imposed by inactive circuits. Critically, within hours of
rewarming and arousal, this extensive tau phosphorylation is reversed, synapses
regenerate rapidly, and cognitive functions are restored without apparent
damage (Popov & Bocharova, 1992; von der Ohe et al., 2006).
Tau
is a protein found in neurons, especially in the long axons that carry signals
between brain cells. Its main job is to stabilize microtubules, which are like
tiny tracks that move nutrients and cell parts around inside the neuron. Think
of tau as railroad ties that keep the neuron’s internal tracks straight and
working efficiently. In Alzheimer’s disease tau becomes
hyperphosphorylated, meaning it gets too many phosphate molecules stuck onto
it. This changes its shape and causes it to fall off the microtubules. Without
tau, the tracks collapse. Worse, the tau proteins start sticking together and
form tangles inside the neurons. These neurofibrillary tangles disrupt
communication, block nutrient transport, and eventually kill the neuron. So, in
AD, tau goes from being a support structure to being a toxic clog.
Fascinatingly,
hibernating, torpid, and starving mammals also show changes in tau—but they
don’t develop Alzheimer’s. In hibernators, like ground squirrels and bears, tau
also becomes hyperphosphorylated. The tau even detaches from microtubules and
there is partial axonal collapse, just as in AD. The railroad tracks are
disabled. In other words, this very expensive process of transporting resources
from the cell body down the axon to the terminal and synapse is frozen. It is
an easy, elegant and reversible way for the brain to save energy. As in AD it
reduces the expensive shuttling of cellular products in the most
energy-expensive areas of the brain, the cerebral cortex and hippocampus.
Fascinatingly, hibernation, prolonged starvation, and Alzheimer’s disease (AD)
all flip tau into an energy-saving configuration by
tagging exactly the same amino-acid sites, those detected by
the AT8 antibody (Ser 202/Thr 205) and by antibodies to Ser
396/Ser 404. Whether it’s a ground squirrel entering torpor, a human fasting,
or a neuron sliding into Alzheimer’s disease, the identical phospho-sites
and writer/eraser enzymes are engaged.
Normally,
many components are shuttled down axons including synaptic vesicles full of
neurotransmitters, mitochondria to power the synapse, mRNA and ribosomes for
local protein synthesis, as well as growth and repair materials for distant
parts of the neuron. Removing the supportive tau freezes activity placing the
neurons in a lean, thrifty suspended state. This starts as soon as hibernation
begins or after several days of food restriciton. For example, it appears in
rat/mouse cortex & hippocampus after 2–5 days of fasting. It causes the
neuron to enter a low-power mode—like pausing a supply chain to
save fuel during a winter shutdown. When this happens, the synapses temporarily
go dark just like a railroad depot in a snowstorm where all the workers have
gone home.
However,
in hibernating and starving mammals, the tau changes are reversible without
lasting repercussions. When hibernating animals wake up the tau phosphorylation
reverses and the tau reattaches to the microtubules reinstating axonal
transport. However, in Alzheimer’s, tau is hyperphosphorylated, but never
de-phosphorylated. The shutdown becomes chronic and the cells never “wakes up.”
Eventually, tau forms tangles, which do not happen in hibernation, causing
permanent destruction.
GSK-3β
is the main tau-phosphorylating enzyme. It adds phosphate groups to tau at many
different sites. In hibernation and starvation GSK-3β becomes transiently
active, but shuts off when the animal rewarms or eats. In Alzheimer’s, the same
enzyme, GSK-3β, is chronically overactive. Something similar is true for the
other tau kinases like CDK5. Also, the enzyme that removes phosphate groups
from tau, PP2A, is reduced in hibernation/starvation and also reduced in
Alzheimer’s.
So,
axonal transport is metabolically expensive because it uses ATP-hungry motor
proteins (i.e. kinesin, dynein) to carry substances up to 1 meter in long
axons. Constant delivery of organelles and materials is required to keep the
synapse alive. By detaching tau and halting the supply chain, the brain can
shut off one of its most expensive processes. In hibernation and starvation,
it’s a reversible suspension of the railroad tracks. In Alzheimer’s, it’s more
like a permanent derailment.
2.2
Amyloid-β Production as a Way to Reduce Energy Expenditure
A
misfolded protein is a protein that bends into the wrong shape. Instead of
floating freely or doing its job, it sticks to other misfolded proteins and
starts to form clumps or aggregates. These can block nutrient flow and
communication. Amyloid is a type of misfolded protein that forms insoluble,
sticky fibers. In Alzheimer’s, the key amyloid protein is called amyloid-beta
(Aβ) but it also plays a role in starvation and hibernation.
Multiple
studies—especially in hibernating ground squirrels, Syrian hamsters, and
bears—have shown that during hibernation levels of amyloid precursor protein
(APP) increase in the brain. It comes from the same source as in AD, improperly
processed or cleaved APP proteins. However, they do not accumulate to the point
of forming amyloid plaques because they are cleared during arousal from
hibernation or after refeeding. So, hibernators appear to intentionally use
amyloid-beta, but their brains manage it carefully, avoiding the pathology seen
in Alzheimer's. Today, researchers believe that Aβ plays an adaptive role in
hibernation and starvation, by suppressing synaptic activity and its energetic
demands. Further, creating these Amyloid proteins is easy and cheap because it
uses preexisting parts (APP).
Synapses
(the connection points between neurons) are one of the largest energy sinks in
the nervous system. When an animal faces several days without food (starvation)
or months without feeding (hibernation), it uses amyloid-beta to slash that
budget. Amyloid-β quiets synapses by dampening their energy-expensive
mechanisms. This includes acetylcholine receptors which buttress learning and
memory. But it also includes NMDA and voltage-gated Ca²⁺ channels which manage
excitatory transmission, conserving vast amounts of energy otherwise used in
signaling and ion pumping. So in effect, this production of amyloid is like
stalling things at the factory, stopping critical junctures for workflow. You
could say that amyloid-beta acts like a lockout supervisor, pausing activity at
the control panels (receptors) of the loading docks (synapses). But let’s talk
more specifically about what it does to acetylcholine receptors.
Acetylcholine
(ACh) is one of the brain’s most important neurotransmitters involved in
attention, learning and memory. It works by binding to special receptors on
neurons, such as the α7 nicotinic acetylcholine receptor (α7nAChR). It’s found
all over the brain, especially in the hippocampus and cortex. When
acetylcholine binds to α7nAChR, the neuron becomes more excitable, synapses
become stronger, and memory circuits become more active and plastic.
Now
here’s the twist: the amyloid-beta protein (Aβ)—especially in its small,
soluble form—can bind to the α7 nicotinic receptor, just like acetylcholine
does. However, amyloid binds much more tightly than acetylcholine and
effectively blocks the receptor (competitive binding), preventing acetylcholine
from working. The receptor is occupied, but it’s not doing its job. When
amyloid-beta clogs up the α7 receptors the brain slips deeper into a
low-activity, energy-saving mode. Thus, in hibernation, torpor, and starvation
acetylcholine activity is intentionally dialed down as a way to save energy.
This supports the idea that Alzheimer’s disease is a pathological version of an
ancient energy-saving response, using the same molecules, same receptors, and
same circuits, just stuck in the on position too long.
Amyloid
formation is not exclusive to the brain—it’s a broader metabolic regulatory
phenomenon. In fact, from an evolutionary standpoint, there’s growing support
for the idea that protein aggregation may serve an energy-saving function under
certain conditions. In type 2 diabetes mellitus (T2DM), amyloid also
accumulates pathologically in the pancreas—specifically in the islets of
Langerhans. But here, it's not Aβ, it's islet amyloid polypeptide (IAPP), also
known as amylin. They form plaque-like deposits that damage or kill beta cells.
This accumulation of IAPP reduces insulin secretion—effectively contributing to
insulin insufficiency and hyperglycemia and in turn reducing glucose uptake by
tissues and accomplishing energy conservation. Amylin fibrils might have
evolved as a self-limiting brake on insulin secretion.
What
emerges is a picture of amyloid aggregation as part of a broader, conserved
strategy to reduce energy expenditure during prolonged energy stress. Thus,
rather than being purely toxic junk, amyloid in both the brain and pancreas
might reflect a thrifty design that goes awry when unregulated—especially in
the context of modern environments (over-nutrition, prolonged lifespan,
sedentary living). It also suggests that therapeutic strategies used in
T2DM—like insulin sensitizers (GLP-1 agonists, SGLT2 inhibitors)—may have
shared relevance in AD, which, as we have discussed, is exactly what recent
clinical trials are now exploring.
In
the evolutionary literature, type 2 diabetes is often interpreted as the
pathological extension of a once-adaptive “thrifty” genotype or phenotype—one
that evolved to conserve energy during periods of caloric scarcity. Traits such
as insulin resistance, fat storage, and reduced glucose uptake would have
conferred survival advantages in ancestral environments marked by intermittent
famine. The recent characterization of Alzheimer’s disease as “type 3 diabetes”
reinforces this perspective, highlighting its shared features with systemic
metabolic disorders—particularly insulin resistance, impaired glucose
utilization, and amyloid accumulation—further supporting the view that
Alzheimer’s, like type 2 diabetes, may represent the maladaptive persistence of
an ancestral energy-conservation program.
2.3
Kinase/Phosphatase Switch (GSK-3β ↔ PP2A)
The
remarkable reversibility in hibernators hinges upon a tightly regulated
biochemical switch involving kinase and phosphatase enzymes that control tau
phosphorylation. GSK-3β (Glycogen Synthase Kinase 3 beta) is a primary kinase
responsible for phosphorylating tau at multiple sites (including AD-relevant
ones like Ser396 and Thr231). It is activated in both hibernation and
Alzheimer’s. During torpor, GSK-3β activity rises, leading to reversible tau
phosphorylation. In AD, persistent GSK-3β activation leads to chronic,
irreversible tau aggregation.
PP2A
(Protein Phosphatase 2A) is the main tau dephosphorylating enzyme in the brain.
It is suppressed during torpor, allowing tau phosphorylation to proceed. In AD,
PP2A is chronically downregulated or inhibited, preventing tau clearance and
promoting neurofibrillary tangle formation. During arousal from torpor,
norepinephrine (from the locus coeruleus) and thyroid hormones suppress GSK-3β
and stimulate PP2A. This rapidly removes phosphate groups from tau, thus
restoring normal neuronal cytoskeletal integrity (Arendt & Stieler, 2003).
In early AD, however, norepinephrine-producing neurons in the locus coeruleus
are among the first to degenerate, which may block this arousal-like reversal
pathway, leading to sustained tau phosphorylation.
2.4
Norepinephrine, Thyroid, and Brown Adipose Tissue (BAT) Arousal Burst
The
rewarming phase in hibernating mammals involves a critical metabolic burst
orchestrated largely by norepinephrine (NE) from the locus coeruleus, thyroid
hormone release, and brown adipose tissue (BAT) thermogenesis. This coordinated
burst re-energizes neurons, rapidly restores ATP availability, and reactivates
metabolic enzymes necessary for neuronal recovery and synapse regeneration.
Interestingly, this NE-mediated metabolic burst precisely mirrors pathways
disrupted early in Alzheimer's disease, providing an additional therapeutic
target for reversing Alzheimer's pathology (Cannon & Nedergaard, 2004;
Tupone et al., 2013).
2.5
Daily Torpor & Synthetic Torpor in Non-Hibernators
Non-hibernating
species, including certain rodents and primates, demonstrate shorter bouts of
daily torpor, during which similar transient phosphorylation of tau, metabolic
reduction, and synapse pruning occur. Importantly, researchers have
experimentally induced synthetic torpor states in laboratory rodents using
pharmacological agents like adenosine A₁ receptor agonists. These animals also
experience profound yet fully reversible tau phosphorylation, synaptic pruning,
and metabolic slowdown, confirming that torpor-like mechanisms are broadly
conserved across mammals, including species closely related to humans (Cerri et
al., 2013; Tupone et al., 2013).
2.6
Insulin and Leptin Receptors — the Body’s Fuel Sensors
During
starvation or hibernation, both insulin and leptin drop, signaling to the
brain: “There’s no food coming in—conserve energy!” This message turns off
systems that use lots of energy (like thinking and moving) and shifts the body
into a low-power mode. In Alzheimer’s, the brain stops responding properly to
insulin and leptin—even when they’re present. This is called resistance. The
result is similar to starvation: reduced brain energy, memory loss, and cell
damage. So the brain ends up acting like it's starving, even though food is
available.
When
leptin and insulin fall, a part of your brain called the hypothalamus activates
certain neurons that release: AgRP (Agouti-related peptide) and NPY
(Neuropeptide Y). These are hunger-triggering signals that also lower body
temperature, reduce activity, and slow metabolism—traits seen in both
hibernation and Alzheimer’s.
2.7
AMPK and mTOR — the Cellular Fuel Gauges
Inside
cells, two important proteins track energy use: AMPK and mTOR. AMPK turns on
when energy is low; it tells cells to slow down, recycle parts, and survive
with less. The other is mTOR which works when energy is high; it tells cells to
grow and make new proteins.
When
animals hibernate or fast, AMPK increases and mTOR decreases—this slows the
brain down and saves energy. In Alzheimer’s, this same balance is off—AMPK
stays high, mTOR stays low. That keeps brain cells in a “shutdown mode” for too
long, which might contribute to shrinkage and memory loss.
2.8
Adenosine Receptors — the Brain’s Dimmer Switch
Adenosine
builds up in the brain the longer you're awake. It tells your brain, “You’re
tired, slow things down.” It acts mostly through the A1 and A2A receptors to
suppress brain activity and encourage sleep or rest—also helpful during torpor
or hibernation. In Alzheimer’s, adenosine signaling becomes too strong or
unbalanced, leading to constant suppression of memory circuits and increased
inflammation.
2.9 Inflammatory
Receptors — IL-1 and TNF
IL-1
and TNF are molecules that trigger inflammation. They bind to receptors that
help the brain respond to stress or infection. In short bursts, like at the
start of torpor, they help animals cool down and conserve energy. But in
Alzheimer’s, these signals become chronic, leading to overactive immune cells
in the brain and destruction of healthy neurons.
1.10 Microglial
Receptors — the Brain’s Cleanup Crew
Microglia
are the brain’s immune cells. They clean up damaged parts, including old
synapses. They use receptors like TREM2 and CR3 to find what to remove. In
healthy development (and especially in hibernation), microglia trim unneeded
connections to make the brain more efficient. But in Alzheimer’s, the cleanup
goes too far. These receptors stay turned on, and microglia start removing
healthy synapses, leading to memory loss.
Temporary Conclusion
These
molecules and receptors aren’t just involved in Alzheimer’s—they’re part of an
ancient survival system: When food is scarce or it’s winter, they help animals
slow their brains, prune unnecessary circuits, and conserve energy. In
Alzheimer’s, these same systems get turned on—but don’t turn off. The brain
ends up stuck in a thrifty, low-power state, unable to recover. This may
explain why Alzheimer’s looks so much like a hibernation program—it uses
precisely the same switches and circuits, but without an exit ramp.
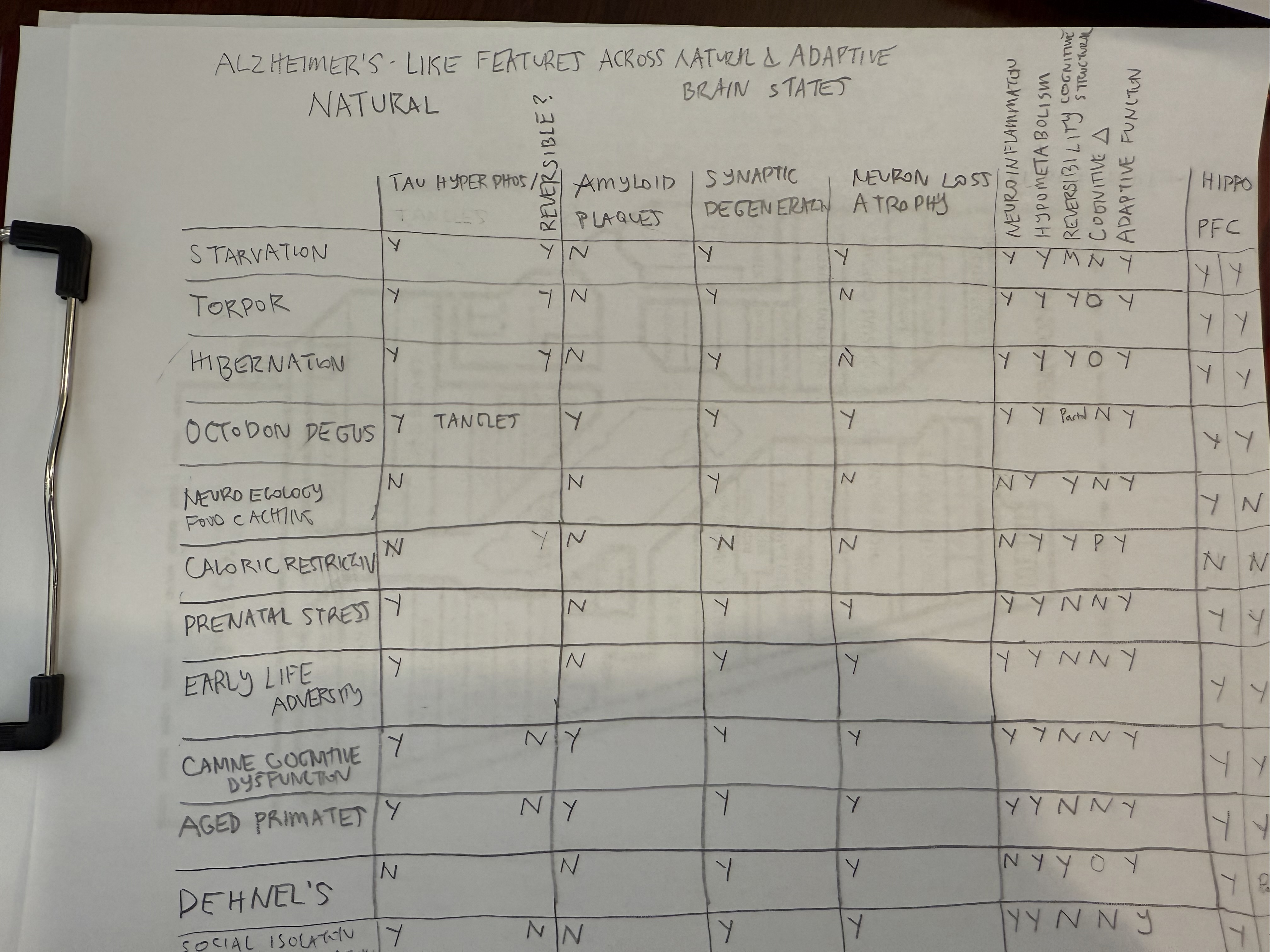
The
next two tables document which natural states demonstrate AD-like pathology.
Alzheimer's Like
Features Across Natural and Adaptive Brain States:
.png)
.png)
3. Mammals with Spontaneous Amyloid + Tau Pathology
3.1
Cetaceans (Dolphins, Whales)
Cetaceans,
such as bottlenose dolphins and pilot whales, exhibit naturally occurring
Alzheimer's-like pathology, including amyloid-beta plaques and tau tangles.
These neuropathological features correlate strongly with extended lifespan,
substantial cortical mass, and metabolic stressors inherent to prolonged diving
and high cognitive demands (Gunn-Moore et al., 2018; Davis et al., 2019).
Importantly, despite prominent pathology, cognitive function in these species
remains largely intact, suggesting effective endogenous mechanisms that limit
functional impairment.
3.2
Great Apes, Dogs, and Cats
Chimpanzees
and other great apes, with lifespans approaching human longevity, display
similar amyloid and tau accumulation patterns in aging brains. These AD-like
features emerge predominantly in associative cortices and hippocampal regions
analogous to those affected early in humans. Yet again, these changes typically
appear only at extreme ages beyond typical reproductive periods, supporting the
hypothesis that metabolic conservation mechanisms only become pathological when
extended unnaturally (Rosen et al., 2008).
Similarly,
aged domestic dogs exhibit Alzheimer's-like (neuritic Aβ plaques) plaques, some
p-tau but rarely full tangles, and cognitive dysfunction, again emphasizing the
role of lifespan extension and metabolic demands in pathology manifestation
(Inestrosa et al., 2005; Head, 2013). Aged cats exhibit extracellular Aβ and
AT8-positive intraneuronal p-tau accumulate with age; cerebral amyloid
angiopathy is also reported. Rhesus macaques show Aβ plaques, tau
phosphorylation, gliosis, synapse loss by around 25 years of age.
3.3
Octodon degus
The
rodent Octodon degus, has several adaptations to its semi-arid
environment in the matorral ecoregion of central Chile. These adaptations, such
as a tendency toward the metabolic syndrome, help it save energy. Many
individuals spontaneously develop classic AD neuropathology, including amyloid
plaques, tau tangles, neuroinflammation, neuron and synapse loss, and cognitive
impairment, closely paralleling human Alzheimer's. The rodent lives 7–9 years at least twice as long as most lab rodents indicating that this
could be an example of late life runaway thrift. They practice corprophagy,
ingesting their feces to extract more nutrients particularly when food is
scarce or low in nutrients. They have a specialized digestive system that
allows them to efficiently extract nutrients from plant matter including dried
vegetation and bark. They have highly reduced basal metabolic rate compared to
other rodents. Their metabolism can vary seasonally with lower rates during the
summer months (nonbreeding season). This comparative model underlines the
evolutionary conserved nature of AD-like responses under metabolic strain.
The
presence of Alzheimer’s-like neuropathology in Octodon degus, which
does not hibernate, strongly supports the idea that AD may be an evolutionary
thrifty program gone awry. In fact, degus are one of the clearest animal cases
showing how normal, adaptive energy-saving responses in the brain can slide
into chronic degeneration under modern or mismatched conditions. Octodon
degus bridge the gap between adaptive plasticity and maladaptive
degeneration. Their spontaneous development of AD-like features in old age
doesn’t just fit the thrift hypothesis—it embodies it. They
show that Alzheimer’s may be less about the “collapse of the brain,” and more
about a strategy for conserving it.

3.4
Exception Species (Naked Mole-Rat) and Lessons in Resilience
A
notable exception to typical mammalian patterns is the naked mole-rat (Heterocephalus
glaber), which despite extraordinary longevity (over 30 years) and high
brain amyloid-beta burden, does not develop pathological plaques, tau tangles,
or cognitive impairment. This remarkable resilience may reflect unique
molecular adaptations to metabolic and oxidative stress, highlighting
protective pathways that prevent the pathological progression of Alzheimer’s
features. Understanding these mechanisms could inspire novel therapeutic
strategies to confer similar protection in humans (Edrey et al., 2013).
3.5
Dehnel’s Phenomenon and Seasonal Brain Size Reduction
Common
shrews (Sorex araneus), unable to store significant fat reserves or
enter true hibernation, employ a unique seasonal survival strategy known as
Dehnel’s phenomenon. Each winter, these shrews undergo substantial brain and
skull size reduction (approximately 20%), significantly lowering metabolic
demands. Remarkably, brain structures regrow during the warmer months,
suggesting innate neural mechanisms capable of reversible brain atrophy and
synaptic remodeling. Such extreme, yet reversible, neural adaptations in a
non-hibernating mammal further support the plausibility of a programmed,
reversible metabolic reduction response in humans. Whether tau participates in Dehnel’s phenomenon remains an
open—and testable—question.
3.6
Relation to Neuroecology in Food-Caching Animals
Across
a surprising range of taxa, from small mammals to food caching birds, animals
trim the most energetically expensive part of their forebrain, the hippocampus,
when heightened spatial memory is no longer worth its metabolic cost. These
seasonal contractions deliver a measurable saving in ATP demand—exactly when
food is scarce or ambient temperature drives up thermoregulatory costs.
Alzheimer’s changes begin in the hippocampus. Alzheimer’s disease even targets
the same hippocampal subfields first, with FDG-PET showing an early, selective
fall in glucose use and synaptic activity. The convergence suggests that the
hippocampal atrophy and hypometabolism seen in AD may represent a deeply
conserved neuro-ecological program that many birds and mammals activate only
temporarily. Understanding how chickadees and shrews switch this circuitry off
and back on each year could therefore illuminate new ways to “wake” the human
hippocampus from the chronic low-power state characteristic of early
Alzheimer’s. Whether tau participates in these phenomena is
also an open question.
The
widespread presence of Alzheimer-like neuropathological responses across
diverse mammalian lineages suggests deep evolutionary roots of metabolic
downregulation programs.
Animal Species that Age into Alzheimer-like Neuropathology
Naturally
|
Taxonomic
group |
Species
/ common name |
Key
Alzheimer-like features reported in aged individuals |
|
Companion mammals |
Domestic dog (Canis familiaris) |
Diffuse → neuritic Aβ plaques,
cerebral amyloid angiopathy (CAA), mild tau-P, Canine Cognitive Dysfunction
that maps to lesion load |
|
Domestic cat (Felis catus) |
Cortical Aβ plaques, AT8-positive
tau neurites, CAA, spatial & social memory loss |
|
|
Rodents |
Octodon degus |
Aβ plaques & CAA, AT8 / PHF-1
tau tangles, microglial activation, object‐ and maze-memory decline |
|
Guinea-pig (Cavia porcellus) |
Human-identical Aβ sequence;
cortical plaques and CAA with age or high-cholesterol diet; sparse tau-P |
|
|
Gray short-tailed opossum (Monodelphis domestica) |
Soluble Aβ accumulation and
learning deficits by mid-life (plaques rare) |
|
|
Naked mole-rat |
High soluble Aβ and tau-P
but no plaque or tangle aggregation → a “resilience”
comparator |
|
|
Small primates |
Gray mouse lemur (Microcebus murinus) |
Aβ plaques, AT8 tau threads +
tangles, gliosis, progressive spatial-memory loss by 4–5 y |
|
Large primates |
Chimpanzee & occasionally gorilla |
Mature Aβ plaques +
neurofibrillary tangles in cortex/hippocampus of >35 y specimens |
|
Rhesus macaque, cynomolgus
macaque, vervet (African-green) monkey |
Age-dependent cortical plaques,
diffuse tau-P, synapse loss, measurable executive-function decline |
|
|
Marine mammals |
Beaked whales, pilot whales,
bottlenose & Atlantic spotted dolphins |
Dense-core Aβ plaques, CAA, AT8 /
PHF-1 tau tangles, microgliosis; strandings often show severe pathology |
|
Other |
Sheep (rare reports), deer (sporadic) |
Focal cortical plaques ± tau-P —
data limited; not yet mainstream models |
Comparing
the living apes provides a living gradient—from mild Aβ accumulation to plaque
+ tangle formation—helping researchers pinpoint where the adaptive
shutdown spirals into irreversible degeneration. All three apes produce
human-sequence Aβ and deposit plaques as they age, but tau activation
advances only as far as each species’ protection/clearance systems allow:
Extent of Alzheimer-like Neuropathology in the Great Apes
|
Feature |
Orangutan
(Pongo spp.) |
Gorilla
(Gorilla spp.) |
Chimpanzee
(Pan troglodytes) |
|
Oldest brains examined |
Captive adults ≈ 28-36 y (late 30s
≈ human 80s) |
Zoo/wild adults ≈ 35-55 y |
Sanctuaries & zoos ≈ 35-61 y
(late 50s ≈ human 90s) |
|
Amyloid-β (Aβ) plaques |
Sparse, diffuse cortical plaques; strong bias toward the
less-sticky Aβ40; meningeal & white-matter CAA small. |
Numerous plaques in association cortex > hippocampus; extensive
CAA (vessel deposits), especially in males. |
Abundant dense-core & diffuse
plaques in cortex and hippocampus;
CAA common; Aβ42 proportion similar to humans. |
|
Tau hyper-phosphorylation (AT8,
PHF-1) |
Largely absent—no
neuronal pretangles, no glial tau. |
Present but limited: AT8-positive astrocytic “coiled bodies,” occasional
neuritic clusters; neuronal pretangles rare; no mature tangles. |
Robust: AT8-positive pretangles, neuritic threads,
astrocytic & oligodendroglial tau; true neurofibrillary tangles in
hippocampus and temporal cortex of the oldest animals. |
|
Neuroinflammation |
Little reported; microglia
quiescent. |
Moderate microgliosis around
plaques and CAA; sex differences noted. |
Pronounced microglial activation
and astrocytosis around plaques and tau inclusions, mirroring human AD
tissue. |
|
Neuron / synapse loss |
Not detected; cortical thickness
preserved. |
No quantified neuron loss; mild
synaptic pruning possible but unproven. |
Subtle neuron loss in entorhinal
cortex and CA1 reported in oldest individuals; synaptic protein reductions
parallel plaque load. |
|
Behavioural / cognitive data |
No systematic late-life testing
(orangutans seldom reach ≥40 y in captivity). |
No formal cognitive batteries;
anecdotal slowing only after mid-40s. |
Documented decline in social
decision-making, memory, and problem-solving after ~45 y; parallels lesion
burden. |
|
Overall “Braak stage” analogue |
Pre-Braak: Aβ present, tau cascade
not triggered. |
Braak I–II: Aβ plus initial tau-P,
but tangles rare. |
Braak III–IV: Aβ + tangles in
limbic & association cortex—closest non-human match to early/mid human
AD. |

3.7
Ancient Torpor Machinery in Placental Mammals: Why Humans Lost Behavioral
Hibernation but Retained Cellular Toolkit
Humans
probably descend from hibernating ancestors. Evidence suggests early mammals
could enter torpor or hibernation-like states. Many modern mammals—especially
small insectivores like shrews, hedgehogs, and tenrecs—exhibit daily torpor or
seasonal hibernation, and are considered evolutionary relics of early mammalian
forms. The molecular machinery required for torpor (e.g. tau phosphorylation,
synaptic pruning, PP2A modulation, thyroid hormone control) is conserved across
nearly all mammals, including humans—even though we don’t behaviorally
hibernate. Certain strepsirrhine primates, like the fat-tailed dwarf lemur (Cheirogaleus),
do hibernate—sometimes for months at a time—with all the classical features:
metabolic suppression, tau phosphorylation, reversible synapse loss. They have
“fat tails” because they store fat in their tails.
Humans,
though not true hibernators, retain a remarkably conserved cellular and
molecular toolkit associated with these states, including the
kinase/phosphatase regulatory mechanisms that modulate tau phosphorylation,
adenosine-based metabolic control pathways, and potent proteostatic clearance
systems (Tupone et al., 2013; Cerri et al., 2013). Understanding these
evolutionary genetic trade-offs and the ancient origin of torpor-related
cellular pathways not only provides insight into Alzheimer's pathogenesis but
also opens new therapeutic avenues focused explicitly on reactivating
protective metabolic mechanisms and reversing pathological processes.
4. The High Cost of Thinking: Life-History
Trade-offs and the Concept of Late Life Thrift
The
human brain is notoriously expensive, metabolically speaking. Despite
constituting only about 2% of total body mass, it consumes roughly 20–25% of
the resting metabolic rate (RMR)—a caloric commitment far exceeding any other
organ in the body, and substantially higher than the cerebral metabolic demand
of other primates or mammals (Raichle & Gusnard, 2002). This energetic cost
is predominantly due to cortical neurons, particularly those within associative
areas like the default-mode network, where continuous baseline activity
maintains readiness for cognitive processing (Buckner et al., 2008). Of course,
these are the same areas effected by AD. Crucially, this metabolic
investment is disproportionately allocated to higher-order processing areas of
the cerebral cortex critical for memory, decision-making, and
planning—precisely those areas compromised early in Alzheimer’s Disease (AD)
(Sokoloff, 1999).
From an evolutionary perspective, the
allocation of energy to cognitive functions must balance against the caloric
returns obtained from environmental exploitation. Ethnographic and
anthropological data from modern hunter-gatherer populations such as the Hadza
and Tsimané show a consistent pattern: caloric productivity in humans typically
peaks between ages 30–40 and subsequently declines sharply, dropping
significantly after age 45–50 (Kaplan et al., 2000; Gurven et al.,
2006). In evolutionary terms, the metabolic cost-benefit ratio of
supporting energetically demanding neural networks declines with age, thereby
potentially selecting for mechanisms to reduce unnecessary energetic
expenditures in the aging brain. AD first throttles and
deactivates the highest cost, plastic networks (prefrontal cortex, posterior
cingulate, medial temporal lobes etc.) while sparing sensorimotor regions
essential for day to day survival, mirroring a form of precision triage rather
than an indiscriminate degenerative map.
Life-history
theory predicts that organisms allocate energy strategically across their
lifespan, optimizing reproductive success and survival. Given the intense
metabolic demands of the human brain and the declining returns on cognitive
investments in later life, there is strong evolutionary rationale for a
mechanism to selectively reduce cerebral metabolism after reproductive
maturity. Strategic pruning of neural networks and reallocation of limited
caloric resources toward essential physiological functions could have prolonged
survival and enhanced indirect fitness benefits such as care-giving, knowledge
transfer, and social roles within the community (Hawkes & Coxworth, 2013).
5. Human Evidence for a Built-In Low-Power Mode
5.1
FDG-PET chronology: hypometabolism precedes amyloid and tau pathology
A
growing body of longitudinal neuroimaging evidence reveals that glucose
hypometabolism, detectable via fluorodeoxyglucose positron emission tomography
(FDG-PET), emerges significantly earlier than the appearance of classical
Alzheimer's biomarkers such as amyloid plaques or tau tangles. Studies from the
Alzheimer's Disease Neuroimaging Initiative (ADNI) and the Wisconsin Registry
for Alzheimer's Prevention (WRAP) demonstrate a clear timeline: reduced
cerebral glucose metabolism in specific cortical regions appears up to 10–15
years before cognitive symptoms or detectable amyloid or tau accumulation
(Jagust & Landau, 2021; Mosconi et al., 2008). This observation strongly
suggests that hypometabolism is not merely a consequence of neuronal injury but
rather a potential initiating factor, consistent with the metabolic-reduction
hypothesis.
5.2
Fuel specificity: preserved ketone uptake, insulin resistance ("type 3
diabetes")
Despite
significant glucose hypometabolism, emerging dual-tracer PET studies show
preserved brain uptake of alternative fuels, particularly ketone bodies.
Clinical research demonstrates that while glucose metabolism significantly
diminishes in early Alzheimer’s and mild cognitive impairment (MCI), the
brain’s ability to utilize ketones remains largely intact (Cunnane et al.,
2016). Such fuel specificity implies that the observed metabolic reduction is
not due to generalized mitochondrial dysfunction or neuron death but instead
reflects a selective downregulation of glucose uptake and processing pathways,
akin to the adaptive metabolic shift seen during fasting or hibernation.
This
selective glucose impairment parallels the widely recognized insulin resistance
in Alzheimer's, often termed "type 3 diabetes" (de la Monte &
Wands, 2008). Reduced insulin sensitivity and signaling within the brain
further underscore an evolutionary context wherein neurons selectively restrict
glucose use—possibly an adaptation to prolonged caloric insufficiency or
energetically demanding conditions. This shift resembles the “fuel switch”
that occurs during starvation, torpor, or hibernation, where the brain begins
favoring alternative substrates (primarily ketones) to reduce reliance on
glucose, which becomes scarce or deliberately restricted.
Multiple
randomized controlled trials (e.g. BENEFIC, Henderson et al.) show that
providing ketones exogenously through medium-chain triglycerides (MCTs) or
ketone esters can raise brain ATP production and metabolism in association
cortices, improve memory and executive function, and restore functional
connectivity on PET and fMRI (Croteau et al., 2018; Fortier et al., 2021). If
cognition and metabolic function can be restored with simple fuel repletion, it
supports the idea that many neurons in early AD are not lost, but rather
functionally silenced or idling—consistent with an evolutionarily conserved
energy-saving mechanism.
Table 1. Therapeutic metabolic trials and cognitive outcomes
|
Intervention |
Duration |
Metabolic outcome |
Cognitive outcome |
Reference |
|
MCT ketogenic supplementation |
6 months |
↑ Brain ketone uptake (PET) |
Improved episodic memory,
executive function |
Fortier et al., 2021 |
|
Ketone ester (single dose) |
Acute |
↑ Parietal metabolism/connectivity |
Improved attention, reaction time |
Cunnane et al., 2016 |
|
Intranasal insulin (40 IU/day) |
4 months |
↑ FDG uptake, especially in APOE
ε4 |
Improved memory, cognitive
performance |
Craft et al., 2017 |
|
GLP-1 agonist (liraglutide) |
12 months |
Stabilized glucose metabolism
(FDG-PET) |
Slowed cognitive decline |
Gejl et al., 2016 |
|
Sodium selenate (PP2A activator) |
3 months |
↓ Phospho-tau (CSF) |
Trend towards cognitive
improvement |
Malpas e |
5.3
Ancient Torpor Machinery in Placental Mammals: Why Humans Lost Behavioral
Hibernation but Retained Cellular Toolkit
Humans
probably descend from hibernating ancestors. Evidence suggests early mammals
could enter torpor or hibernation-like states. Many modern mammals—especially
small insectivores like shrews, hedgehogs, and tenrecs—exhibit daily torpor or
seasonal hibernation, and are considered evolutionary relics of early mammalian
forms. The molecular machinery required for torpor (e.g. tau phosphorylation,
synaptic pruning, PP2A modulation, thyroid hormone control) is conserved across
nearly all mammals, including humans—even though we don’t behaviorally
hibernate. Certain strepsirrhine primates, like the fat-tailed dwarf lemur (Cheirogaleus),
do hibernate—sometimes for months at a time—with all the classical features:
metabolic suppression, tau phosphorylation, reversible synapse loss. They have
“fat tails” because they store fat in their tails.
Humans,
though not true hibernators, retain a remarkably conserved cellular and
molecular toolkit associated with these states, including the
kinase/phosphatase regulatory mechanisms that modulate tau phosphorylation,
adenosine-based metabolic control pathways, and potent proteostatic clearance
systems (Tupone et al., 2013; Cerri et al., 2013). Understanding these
evolutionary genetic trade-offs and the ancient origin of torpor-related
cellular pathways not only provides insight into Alzheimer's pathogenesis but
also opens new therapeutic avenues focused explicitly on reactivating
protective metabolic mechanisms and reversing pathological processes.
6. Therapeutic Opportunities Informed by Hibernation Biology
6.1
Re-fuel Strategies (Ketones, Insulin, GLP-1/GIP, SGLT-2)
If
Alzheimer's disease represents an overextended metabolic conservation response,
then therapeutic strategies should first aim to restore balanced fuel supply to
the brain. As discussed, recent evidence strongly supports the use of ketogenic
strategies, intranasal insulin, GLP-1 receptor agonists, and sodium-glucose
co-transporter-2 (SGLT-2) inhibitors to re-establish metabolic flexibility in
early Alzheimer’s (Cunnane et al., 2016; Craft et al., 2017).
Ketone
supplementation, such as medium-chain triglycerides (MCT) or ketone esters,
bypass impaired glucose metabolism by providing alternative fuel to neurons,
rapidly restoring cognitive functions, synaptic plasticity, and metabolic
activity in compromised regions. Intranasal insulin and GLP-1 receptor agonists
(e.g., liraglutide, semaglutide) directly counteract the cerebral insulin
resistance characteristic of Alzheimer's, re-sensitizing neurons to glucose
utilization pathways. Similarly, SGLT-2 inhibitors can modulate systemic
glucose homeostasis, potentially optimizing cerebral metabolism.
6.2
Proteostatic Mechanisms that Allow Safe Reversal in Hibernators
One
remarkable difference between AD and adaptive states such as hibernation lies
in the robust proteostatic mechanisms that permit safe and complete reversal of
pathological changes in the latter. Hibernating mammals effectively manage
protein aggregates through enhanced protein quality-control systems, including
molecular chaperones and autophagic clearance pathways. Cold-shock proteins
such as RNA-binding motif protein 3 (RBM3) are significantly upregulated during
hibernation and cold stress, directly promoting synaptic integrity, tau
dephosphorylation, and neuronal recovery upon rewarming (Peretti et al., 2015).
Moreover,
the dramatic arousal-induced reactivation of protein phosphatases such as PP2A
rapidly reverses tau phosphorylation, enabling swift restoration of normal
neuronal structure and function (Arendt & Stieler, 2003). These protective
mechanisms appear to be muted or impaired in the human Alzheimer’s brain,
suggesting therapeutic opportunities aimed at restoring these endogenous repair
pathways. Namely, understanding that tau hyperphosphorylation, synaptic loss,
and metabolic depression are naturally reversed during hibernation arousal
offers crucial insights for Alzheimer's therapy, highlighting potential targets
to mimic the hibernator’s natural "OFF-switch."
6.2.1
Norepinephrine / LC stimulation
Norepinephrine
(NE) released from the locus coeruleus (LC) during arousal plays a pivotal role
in rapidly reversing tau phosphorylation, reactivating metabolic processes, and
restoring cognitive function in hibernators. Alzheimer's pathology is characterized
by early LC degeneration and reduced NE signaling. Pharmacological strategies
aimed at boosting LC-NE signaling (e.g., atomoxetine, selective norepinephrine
reuptake inhibitors, vagal nerve stimulation) might effectively emulate this
natural arousal mechanism, facilitating metabolic and synaptic recovery (Mather
& Harley, 2016).
6.2.2
PP2A activators (sodium selenate, SAMe)
Protein
phosphatase 2A (PP2A) activation is critical for tau dephosphorylation during
the natural arousal phase in hibernators. Clinical trials employing sodium
selenate, a PP2A activator, have already demonstrated potential benefits,
significantly reducing phosphorylated tau levels and stabilizing cognitive
function in early Alzheimer's patients (Malpas et al., 2023). Similarly,
S-adenosylmethionine (SAMe), another PP2A-enhancing compound, may represent an
additional therapeutic avenue to reverse pathological tau phosphorylation,
mirroring the hibernation reversal mechanisms.
6.2.3
Thermogenic and thyroid mimetics
Hibernators
exit torpor via a robust metabolic burst involving thyroid hormone release and
brown adipose tissue (BAT) thermogenesis. Translating this biological insight,
therapeutic strategies incorporating mild thermogenic stimuli (e.g., infrared
warming, thyroid hormone analogs) may reactivate dormant neural pathways,
reestablish ATP availability, and stimulate neuronal recovery, providing a
novel and biologically coherent approach to Alzheimer's treatment (Cannon &
Nedergaard, 2004).
6.3
Pulse-torpor Approaches and Synthetic Dormancy Compounds
Given
the success of induced torpor in animal models, researchers could explore
synthetic dormancy protocols for Alzheimer’s treatment. Such
"pulse-torpor" approaches might combine pharmacological induction of
temporary metabolic suppression (e.g., via A₁ adenosine receptor agonists) with
controlled rewarming and metabolic stimulation phases (Tupone et al., 2013).
Short, therapeutic cycles of induced dormancy followed by controlled arousal
could facilitate clearance of pathological tau, restoration of neuronal energy
states, and synaptic remodeling—effectively replicating the reversible cycle
naturally used by hibernators.
6.4
Lifestyle Translations: Intermittent Fasting, Cold-hot Conditioning, HIIT
Lifestyle
interventions that replicate elements of ancestral metabolic stress responses
may offer practical, non-pharmacological methods to prevent or reverse early
Alzheimer's pathology. Intermittent fasting and calorie restriction reliably
activate AMPK/mTOR signaling, reduce insulin resistance, and mimic
starvation-induced metabolic adaptations that could re-establish neuronal fuel
flexibility and resilience (Mattson et al., 2018).
Additionally,
temperature-based interventions, such as alternating mild cold and heat
exposure ("cold-hot conditioning"), can activate brown adipose
tissue, release norepinephrine, stimulate thyroid pathways, and promote
neuronal rejuvenation mechanisms observed during natural arousal from torpor.
High-intensity interval training (HIIT), similarly, induces systemic metabolic
shifts and improves insulin sensitivity, potentially restoring cerebral
metabolic balance (Northey et al., 2018).
Table 2: Candidate Interventions Mapped to Hibernator Arousal
Cascade
|
Stage of Arousal Cascade |
Intervention Strategy |
Potential Therapeutic Agents |
|
Metabolic Fuel Restoration |
Ketogenic supplementation; Insulin
sensitizers; GLP-1 agonists; SGLT-2 inhibitors |
MCTs, ketone esters, intranasal
insulin, liraglutide, semaglutide |
|
Tau Dephosphorylation |
PP2A activation; kinase inhibition |
Sodium selenate, SAMe, GSK-3β
inhibitors |
|
NE-BAT Metabolic Reactivation |
NE enhancement; BAT stimulation;
thyroid mimetics |
Atomoxetine, selective
norepinephrine reuptake inhibitors, thyroid analogs |
|
Synaptic Restoration |
Cold-shock proteins; mild
hypothermia; thermogenic activation |
RBM3 upregulation, cold-hot
conditioning, infrared warming |
(Abbreviations: MCT, medium-chain triglyceride; GLP-1,
glucagon-like peptide-1; SGLT-2, sodium-glucose co-transporter-2; PP2A, protein
phosphatase 2A; SAMe, S-adenosylmethionine; BAT, brown adipose tissue; NE,
norepinephrine; RBM3, RNA-binding motif protein 3.)
These
insights from hibernation biology substantially expand our therapeutic toolkit,
reframing Alzheimer's not simply as neurodegeneration but as reversible
metabolic dormancy. Leveraging this evolutionary wisdom provides unique,
promising, and physiologically coherent opportunities for Alzheimer’s
intervention, focusing explicitly on metabolic recalibration, neuronal
reactivation, and synaptic regeneration.
7. Outstanding Questions and Future Research
The
hypothesis that Alzheimer's disease represents an ancestral metabolic
down-regulation program, pathologically overextended in contemporary humans,
opens numerous intriguing avenues for future investigation. Here are four
critical research directions needed to further substantiate this theory,
advance clinical translation, and bridge disciplinary gaps.
7.1
Quantifying caloric savings of the cortical downshift
A
foundational assumption of the metabolic-reduction hypothesis is that selective
neuronal downscaling provides meaningful energy savings. While general
metabolic shifts have been demonstrated via neuroimaging studies, no precise
quantification of the caloric benefits associated with cortical downregulation
in Alzheimer’s has been systematically conducted. Future studies should aim to
integrate detailed metabolic imaging (FDG-PET, ketone PET, arterial spin
labeling MRI) with metabolic chamber experiments and calorimetry methods to
measure exactly how much energy the brain conserves during progressive
neurodegeneration.
A
detailed model quantifying these metabolic savings in relation to real-world
caloric demands would allow researchers to rigorously test evolutionary
predictions and more accurately assess whether the observed energetic
reductions provide a meaningful adaptive advantage. Such quantification could
fundamentally strengthen the metabolic-reduction model's explanatory power.
7.2
Identifying biomarkers of reversible vs. irreversible tau states
The
concept of Alzheimer’s pathology as a potentially reversible metabolic state
raises the critical issue of distinguishing reversible tau phosphorylation
states from irreversible tau aggregation. The ability to reliably identify
individuals with early-stage, reversible changes would allow timely metabolic
interventions before irreversible damage occurs. Current biomarkers (e.g., tau
PET, CSF phospho-tau, plasma markers) do not effectively discriminate between
reversible, hyperphosphorylated tau and irreversible, aggregated tau deposits.
Thus,
future research should focus on developing advanced biomarker panels—combining
phospho-tau species, PET imaging tracers sensitive to aggregated versus soluble
tau states, and markers of synaptic integrity (e.g., neurogranin, SNAP-25)—to
reliably differentiate reversible tau phosphorylation associated with metabolic
states from irreversible pathological aggregation. Such biomarkers would be
indispensable for optimizing intervention timing and patient selection for
metabolic-based therapies.
7.3
Safety and feasibility of torpor-mimetic therapies in humans
Experimental
evidence in animal models demonstrates that controlled synthetic torpor or
transient hypothermia can safely induce tau hyperphosphorylation and subsequent
de-phosphorylation upon arousal. Translating these concepts safely to humans,
however, remains largely unexplored. Crucial safety questions include the
tolerability of mild therapeutic hypothermia or metabolic suppression,
potential cardiovascular and immune implications, and the reversibility of
induced metabolic downshifts in older adults.
Preliminary
feasibility studies, beginning with controlled, short-duration torpor-mimicking
protocols in healthy volunteers, are necessary to evaluate these therapies.
Early-phase clinical trials should incorporate rigorous metabolic monitoring,
cardiovascular and cognitive assessments, as well as detailed biomarker
analyses, to comprehensively establish safety profiles. Demonstrating human
feasibility would mark a critical step toward using evolutionary-informed
metabolic recalibration strategies to halt or reverse Alzheimer's pathology.
7.4
Cross-disciplinary collaborations: neurology × comparative physiology ×
anthropology
The
integrative nature of the metabolic-reduction hypothesis inherently calls for
interdisciplinary collaboration across traditionally isolated research fields.
Effective exploration and translation of this hypothesis require that
neurologists, comparative physiologists, evolutionary anthropologists, and
metabolic specialists come together to bridge knowledge gaps. Neurologists can
offer expertise in clinical disease processes and therapeutic trial design.
Comparative physiologists contribute essential insights regarding reversible
tau biology and metabolic regulation from animal models. Anthropologists,
comparative physiologists and neuroecologists provide data and context
concerning evolutionary life-history traits, aging patterns, and metabolic
adaptations.
Fostering
these collaborative networks would allow for rich cross-fertilization of ideas,
methods, and perspectives—accelerating discovery and facilitating the
development of novel therapeutic paradigms grounded in evolutionary biology.
Strategic funding and institutional support for multidisciplinary research
initiatives could significantly accelerate this promising avenue toward a
comprehensive understanding of Alzheimer's disease. By addressing these
outstanding questions, researchers will substantially clarify the role of
metabolic conservation in Alzheimer's pathogenesis, determine its therapeutic
potential, and potentially reshape our fundamental approach to dementia
prevention and treatment.
7.5
Implication for other Neurodegenerative Diseases
If
we view Alzheimer’s disease as the pathological prolongation of an ancient
energy-conservation program, this framework could potentially be extended to
help explain other neurodegenerative diseases. Each of these conditions appears
to selectively affect brain regions or circuits with exceptionally high energy
demands, suggesting that they too may reflect maladaptive activation of
conserved “thrift” responses to metabolic stress.
In
vascular dementia (VaD), the cortical and subcortical regions most
affected—such as the watershed cortex and deep white matter—are those most
vulnerable to chronic hypoperfusion. Rather than sudden infarction, these areas
may enter a state of prolonged fuel shortage that drives them into a
hypometabolic, low-functioning state. This pattern is strikingly similar to the
early hypometabolism seen in Alzheimer’s, but here triggered by vascular
insufficiency. From this perspective, VaD may represent a vascularly-induced
thrift program, where circuits enter dormancy to survive low perfusion. The
implication is that therapeutic strategies aimed at metabolic re-fueling—such
as ketones, improved blood flow, or insulin-sensitizing agents—might revive
partially idling circuits before they undergo irreversible degeneration.
In
dementia with Lewy bodies (DLB), pathology centers on regions like the
posterior cingulate and visual association cortex, and includes disruptions in
the cholinergic brainstem system. Alpha-synuclein, the principal aggregating
protein in DLB, is known to inhibit synaptic vesicle recycling—an
energy-intensive process. This suggests that alpha-synuclein may function as a
synaptic “brake,” reducing the energy cost of constant synaptic activity.
Behaviorally, DLB includes vivid hallucinations and REM sleep disturbances,
echoing the dream-rich, low-arousal physiology of torpor. If DLB represents a
form of partial, dysregulated metabolic suppression, then interventions that
restore mitochondrial throughput or mimic arousal neuromodulators such as
norepinephrine and acetylcholine could help restore function even without fully
removing alpha-synuclein aggregates.
Frontotemporal
dementia (FTD) presents a somewhat different profile, targeting anterior brain
regions such as the anterior cingulate, orbitofrontal cortex, and anterior
temporal lobes. These areas support social cognition, emotional regulation, and
executive function—all metabolically expensive and reliant on spontaneous
firing and integration. FTD may represent an alternative “special-purpose”
thrift response, where under conditions of chronic stress or energetic crisis,
the brain selectively sheds high-cost social-cognitive circuits rather than
memory-related ones. The behavioral symptoms—disinhibition, apathy, and loss of
empathy—may mirror what happens in animal models when frontal circuits are
pruned to conserve energy. If so, targeted metabolic interventions that support
prefrontal circuits could slow early FTD progression.
Parkinson’s
disease (PD) affects dopaminergic neurons in the substantia nigra, which are
uniquely vulnerable because they engage in continuous autonomous pacemaking and
calcium signaling—processes that impose extreme mitochondrial stress. The
presence of alpha-synuclein aggregates and mitochondrial downregulation in
these cells may reflect a cell-autonomous attempt to reduce energy consumption.
However, this thrift response ultimately leads to insufficient ATP production
and cellular degeneration. If PD represents a chronic misfiring of this
protective strategy, then therapeutic efforts could focus on supporting ATP
generation through ketones or NAD⁺ precursors, buffering calcium loads, or
introducing structured “rest periods” for pacemaking neurons.
Amyotrophic
lateral sclerosis (ALS) primarily affects motor neurons with extremely long
axons, which must sustain high-speed axonal transport and constant energy
delivery across distances greater than one meter. These neurons are profoundly
dependent on ribosomal translation and mitochondrial transport. In ALS, TDP-43
pathology suppresses protein synthesis and ribosome biogenesis—an energy-saving
adaptation that may become catastrophic over time as motor axons run out of
supplies. From this standpoint, ALS reflects an attempt to downscale
energetically unsustainable axonal function. Restoring mitochondrial
trafficking and supplying alternative fuels like ketones directly to axons may
offer therapeutic benefit by converting maladaptive chronic thrift into a
manageable, potentially reversible state.
Across
these disorders, certain unifying predictions emerge. Early hypometabolism
likely precedes protein aggregation in each case, suggesting that interventions
targeting metabolism could delay or prevent structural damage. Neurons in
affected circuits may respond positively to alternative fuels like
β-hydroxybutyrate or lactate, even if aggregates are still present. The role of
neuromodulators such as adenosine, norepinephrine, and thyroid hormones appears
central in regulating transitions between reversible dormancy and irreversible
degeneration. Therapeutic strategies that mimic or modulate these arousal
signals, or that support cyclic metabolic recalibration rather than linear
decline, may prove useful across Alzheimer’s, vascular dementia, Lewy body dementia,
FTD, Parkinson’s, and ALS.
Ultimately,
this energy-based framing shifts the focus away from protein removal and toward
restoring the brain’s ability to enter and exit metabolic conservation states
in a controlled manner. These diseases may not reflect total failure, but
rather ancient neural responses to stress—programs meant to protect and
preserve neurons under energetic duress, now running open-loop in modern human
lifespans. The challenge and opportunity lie in understanding how to guide
these circuits out of chronic thrift and back toward functional equilibrium.
Alzheimer's Comparison with other Neurodegenerative forms of Dementia:
.png)
8. Conclusion: Waking the Brain from an Over-Extended Thrift
Mode
Integrating
data across multiple biological scales—ranging from human neuroimaging studies
to comparative animal physiology, molecular signaling pathways, and
evolutionary genetics—we find compelling support for this metabolic-reduction
perspective. Human imaging studies consistently demonstrate early cortical
glucose hypometabolism preceding clinical symptoms and protein deposition.
Comparative biology further reveals striking parallels between Alzheimer’s
neuropathology and the fully reversible brain changes observed during torpor,
hibernation, and acute starvation in numerous mammalian species. Critically,
these adaptive states deploy shared molecular pathways, involving reversible
tau phosphorylation, selective synaptic pruning, amyloid accumulation, and
controlled metabolic downshifts—offering a conceptual and practical blueprint
for therapeutic intervention.
The
central shift advocated by this evolutionary-informed perspective is thus one
of therapeutic strategy. Rather than merely attempting to clear amyloid plaques
or tangles after the fact, a promising alternative involves recalibrating
cerebral metabolism and reactivating the brain’s intrinsic metabolic
"wake-up" pathways. Leveraging hibernation biology, synthetic torpor
induction, ketone-based metabolic interventions, and insulin-sensitizing
therapies represent a new generation of Alzheimer’s treatments, designed not
just to slow or stabilize progression, but potentially reverse pathology by
restoring dormant neural circuits.
This
reframing necessitates collaborative research programs bridging neurology,
comparative physiology, molecular neuroscience, evolutionary anthropology, and
metabolic medicine. Addressing critical questions—such as quantifying energy
savings from neural downshifts, distinguishing reversible from irreversible tau
states, and safely translating torpor-mimetic therapies to humans—holds promise
to transform our clinical approach. Indeed, viewing Alzheimer's through the
evolutionary lens of energy economics not only advances our fundamental
understanding but presents immediate opportunities for novel, targeted, and
potentially transformative interventions aimed at waking the aging brain from
its over-extended thrift mode.

Conclusory Table: New evidence—beyond
the 2009 paper—for framing Alzheimer’s disease (AD) as an over‑extended, brain‑energy‑reduction
program
|
Evidence pillar |
New highlights from our 2024‑25
review |
Why it strengthens the “metabolic‑program”
model |
|
1 Early, fuel‑specific hypometabolism precedes
plaques/tangles |
Longitudinal FDG‑PET shows cortex‑wide glucose use falling
years before tau or amyloid PET turn positive and before cognitive
decline. Hypometabolism predicts who will accumulate tau next. |
Positions reduced energy use as a trigger, not a by‑product,
of pathology. |
|
2 Restoring fuel flow improves cognition |
6‑month MCT (ketogenic) BENEFIC trial → ↑ ketone uptake in
attention network, ↑ functional connectivity, better memory in
MCI ; 15‑month open MCT study stabilised MMSE in 80 % of mild‑AD
participants. |
Shows that supplying alternative “torpor fuel” reverses
part of the deficit—exactly what would help a brain stuck in a thrift mode. |
|
3 Intranasal or sensitiser insulin boosts brain
metabolism |
Multiple RCTs of intranasal insulin report short‑term
memory gains and higher regional FDG uptake in APOE4 carriers. |
Treats the insulin‑resistant, low‑glucose state as a
reversible throttle—matching a programmed reduction. |
|
4 Reversible AD‑type tau in hibernators and
“synthetic torpor” |
Ground‑squirrels, hamsters, bears, and pharmacologically‑torpid
rats show massive tau phosphorylation, synapse loss, hypometabolism
during torpor that clears within hours of arousal via PP2A and NE
bursts. |
Direct demonstration that the molecular signature of AD is
a normal, energy‑saving state that can be switched off. |
|
5 Cross‑species occurrence tracks longevity
& metabolic stress |
Spontaneous amyloid + tau pathology verified in
aged dolphins, chimpanzees, dogs, degus—all long‑lived or prone to
insulin resistance; naked‑mole‑rats accumulate soluble Aβ but never aggregate
or degenerate. |
Shows the program is ancient, turns pathological only
under certain life‑history or metabolic contexts; also proves plaques/tangles
can be benign or reversible. |
|
6 Selective “hit‑map” matches energy triage |
Human and animal data confirm association‑cortex/hippocampus
(highest idle glucose burn) are first to decline, primary sensory–motor areas
are last. |
Aligns perfectly with a strategy that trims the most
energetically costly yet least survival‑critical tissue. |
|
7 Neuromodulator OFF‑switch circuitry is
already perturbed in prodromal AD |
A₂A‑adenosine receptor density is up‑regulated, locus‑coeruleus
NE neurons are lost, orexin and thyroid signals are dysregulated before
dementia. |
Matches the same levers that flip torpor on/off—suggesting
AD brains are stuck mid‑cycle. |
|
8 Thrifty genotypes and metabolic syndrome
overlap |
APOE‑ε4 and other famine‑advantage alleles raise AD risk
but also correlate with enhanced fertility or early‑life survival under
infection/malnutrition. |
Confirms the trade‑off logic: genes that helped conserve
energy under scarcity now predispose to late‑life thrift overshoot. |
|
9 Calorie restriction & intermittent
fasting blunt pathology |
CR / fasting‑mimicking diets in AD mice lower Aβ load,
reduce P‑tau, improve cognition—paralleling natural torpor benefits. |
Manipulating the same metabolic pathways can push the
program back toward adaptive territory. |
|
10 Synthetic torpor rescues AD model mice |
Single pharmacologically‑induced torpor cycle (A₁‑adenosine
agonist) restored LTP and memory in APP/PS1 mice while clearing P‑tau. |
Proof‑of‑concept that intentionally cycling into—and out
of—thrift mode can repair diseased circuitry. |
8998CD7E7D
ReplyDeleteGörüntülü Sex
Sanal Sex
Telegram Show